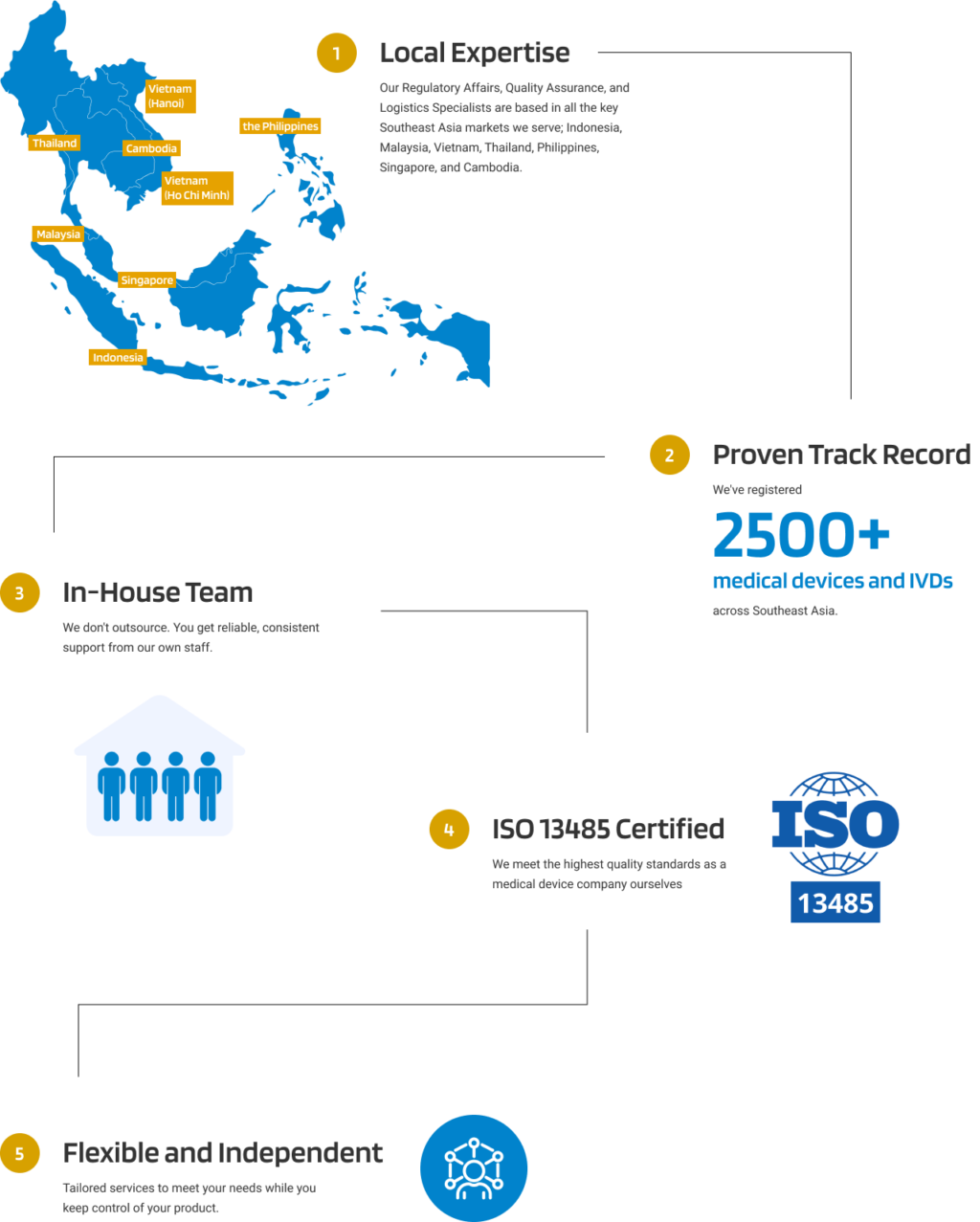
Medical Device Post-Market Surveillance in Indonesia
Why Medical Devices PMS matters in Indonesia
Post-Market Surveillance (PMS) of medical devices in Indonesia is an important regulatory framework designed to maintain the safety, quality, and effectiveness of medical devices after they’ve been approved and released to the market. PMS helps identify, assess, and reduce risks through continuous monitoring, reporting, and corrective action.
At Andaman Medical, we provide comprehensive post-market surveillance solutions tailored to Indonesia’s regulatory framework. Our expertise in local requirements and industry best practices helps you stay compliant, mitigate risks, and uphold the highest standards of product quality.
Here’s how Andaman Medical helps you solve the challenges of medical device post-market surveillance in Indonesia:
Regulatory Compliance
Regulatory Compliance
Post-market surveillance (PMS) is a mandatory requirement by the Ministry of Health to ensure continued safety, effectiveness, and quality of medical devices in the market. Regular monitoring helps maintain compliance with local regulations and avoids regulatory disciplinary measures.
Early Detection of Issues
Early Detection of Issues
PMS allows for the early identification of potential safety concerns, performance issues, or adverse events related to your device. This proactive approach helps prevent widespread problems and protects patients from harm.
Improvement of Product Quality
Improvement of Product Quality
Continuous monitoring provides valuable feedback on device performance in real-world settings. This information can be used to enhance product design, manufacturing processes, and overall quality, leading to better patient outcomes.
Maintaining Market Authorization
Maintaining Market Authorization
Effective post-market surveillance is critical for maintaining your device’s market authorization. Ongoing compliance with surveillance requirements demonstrates your commitment to product safety and helps avoid the risk of losing regulatory approval.
Building Trust with Healthcare Providers
Building Trust with Healthcare Providers
PMS data offers valuable insights into the ongoing performance and safety of medical devices. By systematically monitoring and reporting real-world data, PMS helps healthcare professionals make informed decisions based on up-to-date evidence. This process fosters confidence in your products and supports their safe and effective use in clinical settings.
Risk Management and Mitigation
Risk Management and Mitigation
PMS helps manufacturers identify and address risks associated with their devices, enabling timely corrective actions and reducing the likelihood of costly recalls, legal issues, and damage to brand reputation.
Our Medical Device Post Market Surveillance Services
Report Management
Support for your mandatory reporting of Adverse Effects and Field Safety Corrective Actions
Documentation
Identify requirements in medical device directives, standards, and guidance documents
Data Evaluation
Evaluate your PMS data to ensure that existing processes and outputs are fully compliant
Maintain Compliance
Monitor and report on any regulatory changes to ensure ongoing compliance
What You Need to Know About Medical Device PMS in Indonesia
There are four pillars to the post-market surveillance system in Indonesia, through which medical devices are proactively monitored and sampled by regulatory authorities from the Ministry of Health. These efforts are supported by collaborations with hospitals, healthcare facilities, customs, and provincial health officers. In cases of serious violations, law enforcement agencies may also be involved. These pillars are:
Periodic sampling of medical devices is conducted for laboratory testing to verify compliance with regulatory standards. Any necessary corrective or preventive actions are communicated to the manufacturer through the medical device license holder.
Periodically the MoH will audit production and distribution facilities’ compliance to the Quality Management System
Mandatory reporting of Unexpected Incidents and FSCA cases through the e-Watch system of the Ministry of Health, Indonesia.
The MoH supervises medical device advertising with related stakeholders and issues public warnings if necessary.
Medical device license holders are required to report the following information through the Ministry of Health’s e-Watch system to ensure full traceability of the distribution and location of medical devices:
- Unexpected Incidents (UI), referred to as Adverse Events (AE) in the AMDD
- Field Safety Corrective Actions (FSCA)
- Product Recalls
Andaman Medical Offers Access to
Medical Device E-Watch System
Andaman Medical assists clients in reporting through the e-Watch system, a government platform for comprehensive post-market surveillance. The system facilitates mandatory reporting of Unexpected Incidents (referred to as KTD — Kejadian Tidak Diinginkan in Indonesian regulations), Field Safety Corrective Actions (FSCA), and Product Recalls.
The e-Watch system also enables clients to track early detection alerts and access comprehensive updates on medical devices with potential or identified risks, ensuring timely and traceable responses to uphold high safety and efficacy standards across Indonesia’s healthcare landscape.
Medical Device E-Report System
Our experts are ready to assist you in reporting the distribution of medical devices and household health supplies (PKRT) through Indonesia’s e-Report system. This system ensures comprehensive traceability of medical devices and PKRT within the country, supporting effective post-market surveillance and enabling prompt action in the event of recalls or safety concerns.
Andaman Medical can act as the appointed distributor or license holder, responsible for submitting the required information to Indonesia’s Medical Device e-Report System.

Unexpected Event Reporting
An event has occurred in relation to the use of a medical device.
It has caused serious deterioration/serious injury to the user or others.
It has caused the death of a patient, user, or other person.
It has caused a serious threat to public health with mass impact.
An event could result in the death or serious injury to the user or others if repeated.
Unexpected Incident and FSCA Reporting Timeline
Unexpected Incident shall be reported within:
Within 48 hours:
If the incident or adverse event may cause a threat to public health.
Within 10 days:
If the incident or adverse event causes serious deterioration in health.
Within 30 days:
If the incident or adverse event has the potential to cause death or serious injury.
What are the timelines for Unexpected Event reporting?
48 hours
10 days
10 days
30 days
How Unexpected Incident and FSCA Reporting Works
Field Safety Correction Action (FSCA) is one aspect in the Good Distribution Practice for Medical Device (GDPMD) that must be met by distributors:
Incident Notification
The case of an Unexpected Incident or Adverse Event (AE) is reported to Andaman Medical.
Information Gathering
Andaman liaises with all related stakeholders to obtain complete and accurate information regarding the incident.
e-Watch Reporting
An online report is submitted via the Ministry of Health's e-Watch system according to the required reporting timelines (48 hours, 10 days, or 30 days depending on severity).
Initial Investigation
Initial Corrective & Preventive Actions
Andaman coordinates with the manufacturer to determine and implement Initial Corrective and Preventive Actions to reduce potential risks.
Further Investigation & FSCA Decision
If an unacceptable risk is identified:
Final Report Submission
Andaman prepares and submits the Final Follow-Up Report (including root cause analysis and final investigation results) within 30 days or as required by the Ministry of Health.
Regulatory Feedback
Andaman communicates any feedback from the Ministry of Health to all involved parties, ensuring full traceability and regulatory compliance.
FSCA reporting
Distributors must have standard operating procedures for FSCAs.
Distributors shall assign responsibility for planning, implementing, and reporting corrective actions.
Distributors shall establish product recall procedures after coordinating with producers. If or when a decision is made to withdraw the product, a recall notification must be made.
Distributors must report their planned corrective action activities to the competent authority.
Distributors must inform corrective actions to consumers who have received the product, according to the level of importance of the FSCA.
Records of repair activities must be maintained.
Distributors must have standard operating procedures for FSCAs.
Distributors shall assign responsibility for planning, implementing, and reporting corrective actions.
Distributors shall establish product recall procedures after coordinating with producers. If or when a decision is made to withdraw the product, a recall notification must be made.
Distributors must report their planned corrective action activities to the competent authority.
Distributors must inform corrective actions to consumers who have received the product, according to the level of importance of the FSCA.
Records of repair activities must be maintained.
Adequate corrective actions with respect to the safety and functional aspects of the product shall be determined as soon as possible and implemented in order to eliminate the acute hazard. Necessary actions shall be carried out based on the guidelines from the Principal/Manufacturer and, if applicable, in accordance with instructions from the Ministry of Health (MoH).
Navigating post-market surveillance in Indonesia can be challenging, but Andaman Medical is here to streamline the process and ensure you stay fully compliant with ease. Connect with our experts today and take the first step toward hassle-free compliance!