Medical Device Post Market Surveillance (PMS) in the Philippines
Medical Device PMS Management for Regulatory Assurance and Continued Market Presence
- Philippines
Managing medical device post-market surveillance in the Philippines requires consistent regulatory attention and familiarity with national compliance expectations. Once a device is approved and made available on the market, manufacturers and their appointed local representatives must continue to meet post-market responsibilities under the country’s PMS framework.
Post-Market Surveillance (PMS) in the Philippines establishes the processes for monitoring device performance after commercialization. This includes maintaining complaint records, evaluating reported incidents, fulfilling reporting obligations, and implementing corrective actions in accordance with national regulatory requirements.
At Andaman Medical, we oversee these post-market responsibilities on your behalf. Our team supports adherence to local regulations, maintains organized documentation systems, and coordinates required reporting activities to help ensure your products remain aligned with the Philippines’ regulatory framework.
Why Medical Device PMS Matters in the Philippines
Post-Market Surveillance (PMS) serves as an important mechanism within the Philippines’ medical device regulatory system once products are placed on the market.
Manufacturers and local representatives are responsible for maintaining complete documentation, reviewing reported concerns, and carrying out required reporting activities under national guidelines.
Here’s how Andaman Medical helps you overcome the challenges of medical device post-market surveillance in the Philippines:
Regulatory Compliance
Regulatory Compliance
PMS is a mandatory requirement by the Ministry of Health to ensure the safety, effectiveness, and quality of medical devices. Regular monitoring maintains compliance and avoids penalties. We manage your PMS activities, ensuring full compliance while mitigating risks of non-compliance and penalties.
Early Detection of Issues
Early Detection of Issues
Post-market surveillance identifies potential safety concerns, performance issues, or adverse events early. This proactive approach prevents widespread problems and ensures patient safety. We conduct monitoring and reporting to identify risks, enabling timely corrective actions to protect patients and your product’s reputation.
Improvement of Product Quality
Improvement of Product Quality
Monitoring device performance in real-world settings provides valuable feedback to improve product design, manufacturing, and quality. These enhancements lead to better patient outcomes and stronger market presence. We analyse PMS data to deliver insights that refine your products and maintain high-quality standards.
Maintaining Market Authorisation
Maintaining Market Authorisation
Effective PMS is essential for maintaining your device’s market authorisation. Meeting surveillance requirements demonstrates your commitment to safety and helps avoid losing regulatory approval. We ensure you meet all PMS obligations, keeping your device compliant and authorized in Malaysia.
Building Trust with Healthcare Providers
Building Trust with Healthcare Providers
PMS data demonstrates ongoing device safety and performance, building confidence among healthcare professionals. This trust leads to increased adoption of your products and stronger relationships. We deliver PMS reports and insights that enhance credibility and strengthen trust with healthcare providers.
Risk Management and Mitigation
Risk Management and Mitigation
Post-market surveillance helps identify and address risks, enabling corrective actions to prevent recalls, legal issues, and reputational damage. We manage risks by identifying issues, implementing corrective measures, and protecting your brand.
Medical Device PMS Requirements in the Philippines
In accordance with Annex 5 of the AMDD, the following is required as part of a PMAS in the Philippines:
Importation and/or distribution records
Complaint records
Adverse Event (AE) reporting criteria and reporting format
Field Safety Corrective Action (FSCA) reporting format
All medical device establishments, manufacturers, distributors, importers, exporters, wholesalers must have a Quality Management System (QMS) and relevant documentation in place. This is checked and audited by the Philippines FDA during its routine inspections. The QMS must include an established Risk Management Plan (RMP) and Standard Operating Procedures (SOPs) together with their related forms for Good Storage and Distribution, Complaint Handling, Product Recalls, Medical Device Disposal/Destruction.
IMPORTANT REMINDER
Adverse Event reporting requirements in the Philippines
| Adverse events | Report within |
|---|---|
| Serious threat to public health: Immediately or not later than 48 hours | 48 hours |
| Death | 10 days |
| Serious deterioration in state of health | 10 days |
| Possible death or serious injury if the adverse event were to recur | 30 days |
- An Adverse Event has occurred
- The device is associated with the AE
- The AE led to one of the following outcomes:
- A serious threat to public health
- Death of a patient, user or other person
- No death or serious occurred but the event that might lead to death or serious injury of a patient user or other person if the event recurs.
As part of PMS for medical devices in the Philippines, the evaluator follows up with the reporting person for further information and processes valid reports using CDRRHR-PRSDD-Form 021. If the suspected medical device is registered, the evaluator issues Corrective and Preventive Action (CAPA) to the distributor, manufacturer, or owner about the adverse event/complaint and requires them to conduct corrective action and submit a report. This may lead to the possible issuing of an Advisory or an Advisory plus surveillance or recall of the suspected device.
Any adverse event occurring outside the Philippines do not qualify for reporting to the PFDA, unless the user is Filipino and the device is obtained from the Philippines, or it is otherwise requested. The country of occurrence should be stated.
Navigating post-market surveillance in Philippines can be challenging, but Andaman Medical is here to streamline the process and ensure you stay fully compliant with ease. Connect with our experts today and take the first step toward hassle-free compliance!
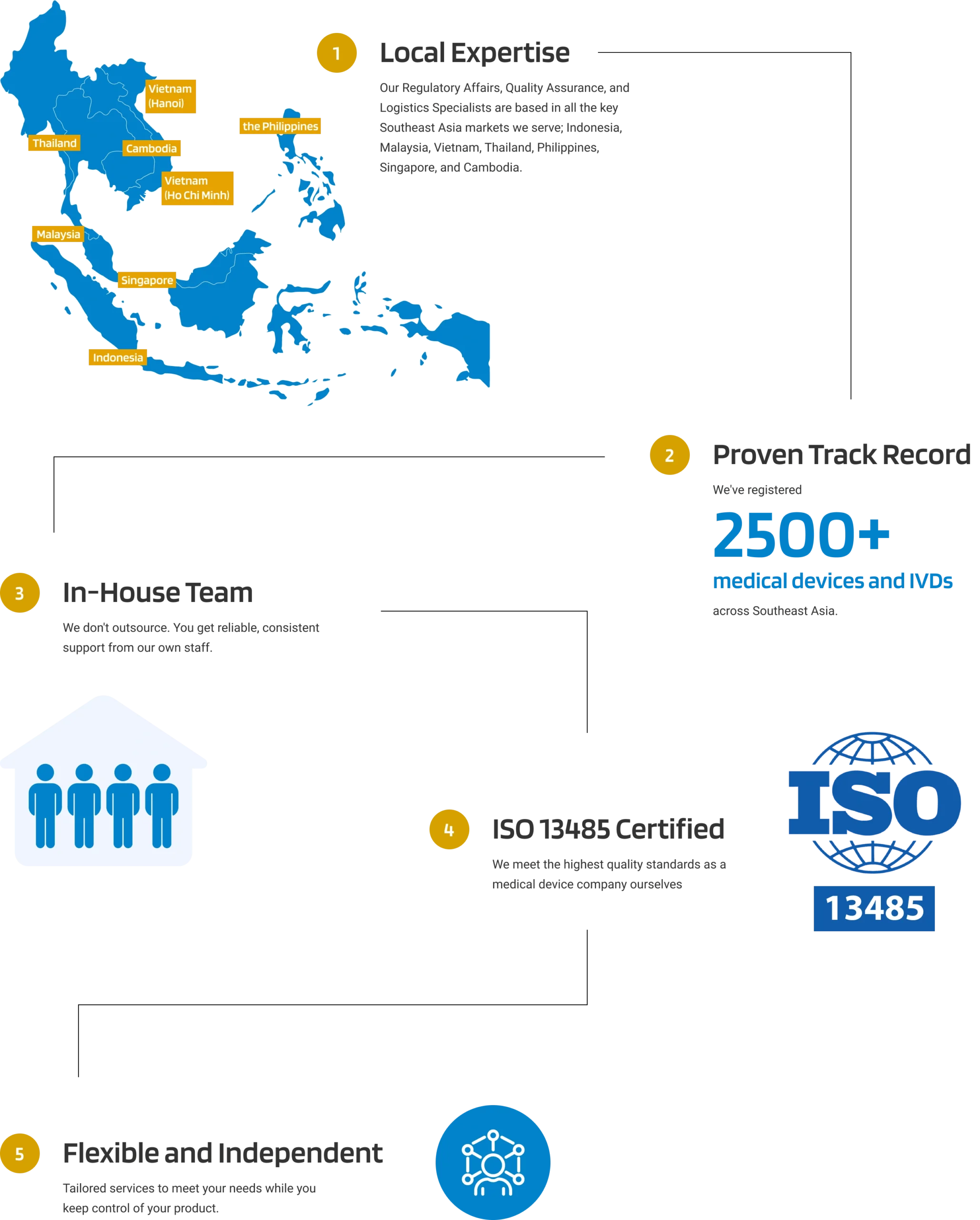
Why Choose Andaman Medical?
Our expert guidance through the region's evolving regulatory landscape ensures your product remains compliant, allowing you to bring your products to market efficiently and effectively.
Experienced In-house Team
across 7 Southeast Asian Countries
Full Control with Seamless
Distributor Switching
Singular Point of Contact
for Registration
QMS Compliance
ISO 13485 Certified
Overcome Language Barrier
With Multilingual Local Team
One-Stop Access
into Southeast Asia
Why Choose Andaman Medical for Your Medical Device Post-Market Surveillance in Philippines
Post-Market Surveillance of Medical Devices in the Philippines
The Philippines’ Food and Drug Administration (PFDA) is the local authority responsible for medical devices in the Philippines and requires all medical device manufacturers and distributors (whether importer, exporter or wholesaler) to undertake post-market surveillance (PMS) and have a PMS plan. The Philippines FDA has chosen to adopt the Post-Market Alert System (PMAS) for Defective Devices of the ASEAN Medical Device Directive (AMDD), please read on to understand how post-market surveillance is implemented in the Philippines.
What are the requirements for medical device PMS in the Philippines?
In accordance with Annex 5 of the AMDD, the following is required as part of a PMAS in the Philippines:
1.
Importation and/or distribution records
2.
Complaint records
3.
Adverse Event (AE) reporting criteria and reporting format
4.
Field Safety Corrective Action (FSCA) reporting format
All medical device establishments, manufacturers, distributors, importers, exporters, wholesalers must have a Quality Management System (QMS) and relevant documentation in place. This is checked and audited by the Philippines FDA during its routine inspections. The QMS must include an established Risk Management Plan (RMP) and Standard Operating Procedures (SOPs) together with their related forms for Good Storage and Distribution, Complaint Handling, Product Recalls, Medical Device Disposal/Destruction.
Please note: In the Philippines an importer is the Local Authorized Representative of Foreign Manufacturers please refer to our Registration of Medical Devices in the Philippines for clarification.
Adverse Event reporting requirements in the Philippines
The following medical device associated AEs must be reported to the Philippines FDA:
| Adverse events | Report within |
|---|---|
| Serious threat to public health | 48 hours |
| Death | 10 days |
| Serious deterioration in state of health | 10 days |
| Possible death or serious injury if the adverse event were to recur | 30 days |
Medical Device Companies reporting Adverse Events will need to send their AE report via email to the Director of the Center for Device Regulation Radiation Health and Research (CDRRHR) and the Division Chief of Product Research and Standard Development Division (PRSDD). The FDA evaluator accepts reports from all distributors, manufacturers, owners, end-users of devices using the prescribed format (CDRRHR-PRSDD-Form 0.20). The FDA evaluator validates any adverse event (AE) which meets the three basic reporting criteria listed below:
- An Adverse Event has occurred
- The device is associated with the AE
- The AE led to one of the following outcomes:
- A serious threat to public health
- Death of a patient, user or other person
- No death or serious occurred but the event that might lead to death or serious injury of a patient user or other person if the event recurs.
The evaluator follows up with the reporting person for further information and processes valid reports using CDRRHR-PRSDD-Form 021. If the suspected device is registered, the evaluator issues Corrective and Preventive Action (CAPA) to the distributor, manufacturer or owner about the adverse event/complaint and requires them to conduct corrective action and submit a report. This may lead to the possible issuing of an Advisory or an Advisory plus surveillance or recall of the suspected device.
Any adverse event occurring outside the Philippines do not qualify for reporting to the PFDA, unless the user is Filipino and the device is obtained from the Philippines, or it is otherwise requested. The country of occurrence should be stated.
How Andaman Medical can help you with post-market surveillance in the Philippines
With 6 local offices throughout Southeast Asia, Andaman Medical provides support for your post-market surveillance activities including adverse event and FSCA mandatory reporting. Our local, in-house staff liaise with the Food and Drug Administration to help you maintain compliance once your medical device is placed on the market. Our services include:
- support for your mandatory reporting of Adverse Events and Field Safety Corrective Actions
- identify requirements in medical device directives, standards, and guidance documents to ensure effective implementation of a post-market surveillance system
- evaluate your PMS data to ensure that existing processes and outputs are fully compliant
- monitor and report on any regulatory changes to ensure ongoing compliance